Variant calling entails identifying single nucleotide polymorphisms (SNPs) and small insertions and deletion (indels) from next generation sequencing data. This tutorial will cover SNP & Indel detection in germline cells. Other more complex rearrangements (such as Copy Number Variations) require additional analysis not covered in this tutorial.
Note: This tutorial uses and older version of GATK (3.x). An updated workflow for variant calling using GATK4 is described here.
Author:
Mohammed Khalfan – mk5636@nyu.edu
Slides:
https://docs.google.com/presentation/d/1Mq7YWGz-PK9myvNl52MHNavK_SYCXWf3GQAUiO6wxlA/edit?usp=sharing
Required Modules
- Bwa 0.7.8
- Picard-tools 1.129
- Gatk 3.3-0
- Samtools 1.3
- Snpeff 4.1
- Tabix 0.2.6 (part of HTSlib)
- IGV
Sample Dataset
prince:/scratch/courses/HITS-2018/variant_calling.tar.gz
dalma:/scratch/gencore/datasets/variant_calling.tar.gz
Introduction
Identifying genomic variants, such SNPs and indels, can play an important role in scientific discovery. Identifying variants is conceptually simple:

But in practice, it can look more like this:

The key challenge with NGS data is distinguishing which mismatches represent real mutations and which are just noise?
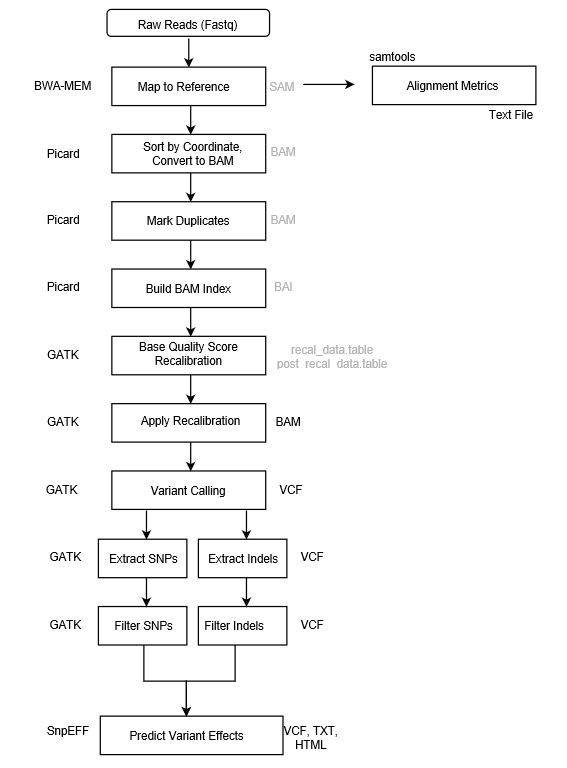
We use the Genome Analysis Toolkit and the best practices for variant discovery analysis outlined by the Broad Institute.
Overview
Each of the steps in the flowchart below is explained within the step-by-step protocols that follow.