Now that we have cleaned data, we still have no idea what we sequenced, though we have an idea of what it is not. Taxonomic classification allows us to assign a rank to each read so that we can obtain a community profile that will open multiple paths to analyses downstream. This is done by querying your dataset against a database that has taxa-specific entities that are associated with a taxonomic identifier.
A nice comparison of classification tools can be found here.
Kraken Taxonomic Classifier
Often when I get a metagenomic dataset I’m very interested to see:
- If we’ve sequenced anything of importance
- What we’ve sequenced
- Proportions of what we’ve sequenced
Answers to these questions are not only important in deciding how to approach your analysis, but can also assess the lab side of things.
Assuming you have no idea what you’ve sequenced, this is generally a good first step as it is easy, reasonably quick, and informative, especially when trying to identify contaminating sequences, which are necessary for contaminant removal. To start, we will use Kraken – a very fast, easy to use taxonomic classifier. This uses the NCBI RefSeq database, represented by a series of clade-specific k-mers which your reads of interested are queried against. By doing this we will get a general idea of what we have in our sample.
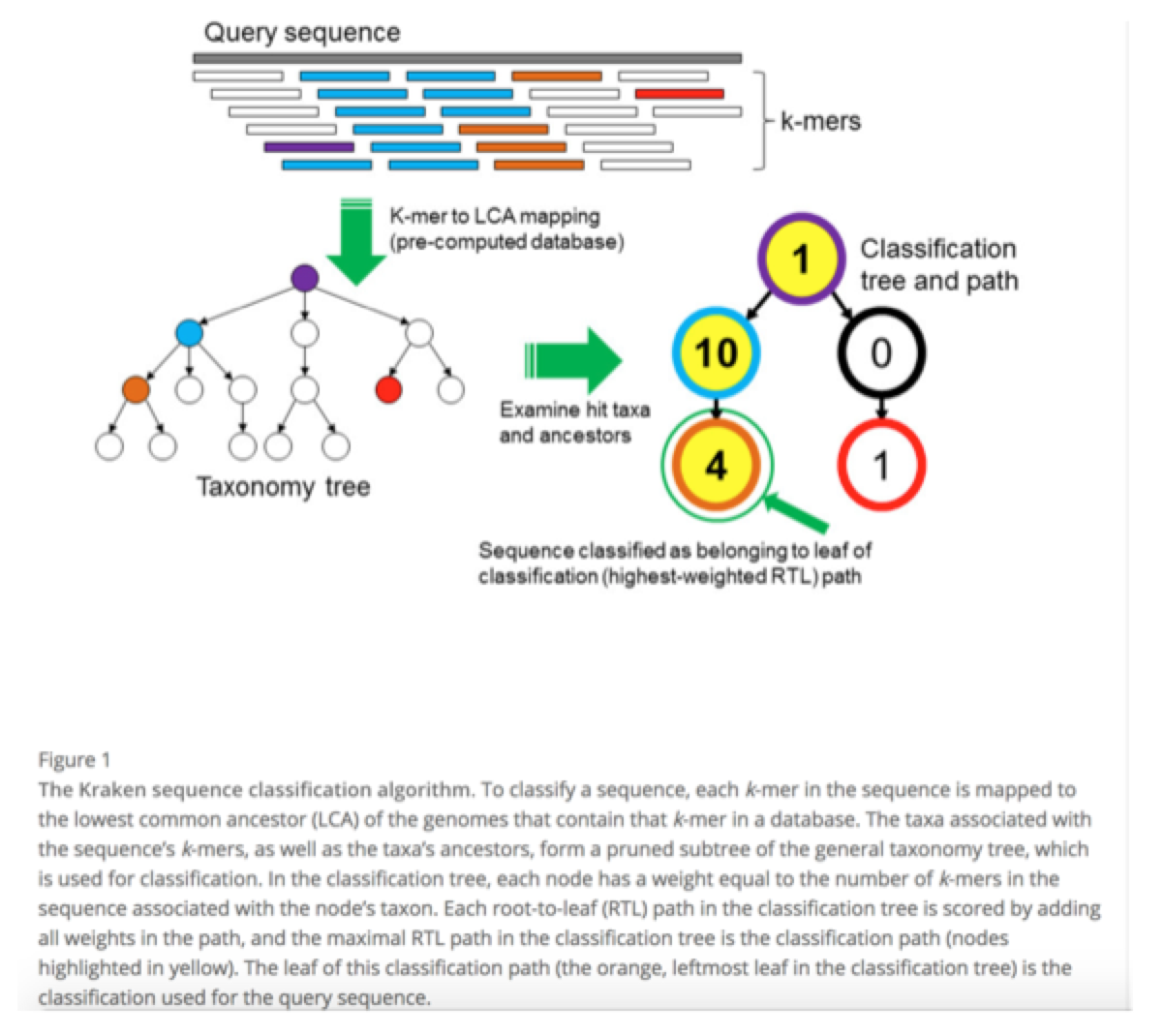
Kraken Databases
Kraken requires a database that stores the information about the known sequences that you’re querying against. A Kraken database is a directory containing at least 4 files:
- database.kdb: Contains the k-mer to taxon mappings
- database.idx: Contains minimizer offset locations in database.kdb
- taxonomy/nodes.dmp: Taxonomy tree structure + ranks
- taxonomy/names.dmp: Taxonomy names
Of these databases, there are two to choose from:
- MiniKraken Database
This is a pre-built 4 GB database constructed from complete bacterial, archaeal, and viral genomes in RefSeq (as of Dec. 8, 2014). This can be used by users without the computational resources needed to build a Kraken database. - Standard Kraken Database
This is the complete NCBI taxonomic information, as well as the complete genomes in RefSeq for the bacterial, archaeal, and viral domain. This is huge – it requires ~62GB RAM to run a search and ~160GB disk space to create.
Alternatively, you can create custom databases and you can find the documentation here.
Obtaining Databases
In the event that you do not have the databases readily available, you can follow these steps. Both of these databases are already available on Dalma, so no need to run these commands.
For the standard Kraken database:
kraken-build --standard --db $DBNAME
$DBNAME can be whatever you want it to be. For clarity, I name it ‘standard’
For the MiniKraken database, you only need to download and decompress it:
wget https://ccb.jhu.edu/software/kraken/dl/minikraken.tgz
tar -xvzf minikraken.tgz
Running Kraken
Now that we have the databases, we can get kraken 😉
Kraken requires two arguments to run:
- A Kraken database – for this tutorial we will use the MiniKraken database.
- A reads file
The full list of parameters can be found in the Kraken user’s manual, but the ones I generally use can be found here:
- ‘–threads‘ specifies how many threads to use, and it makes things much faster.
- ‘–preload‘ this loads the database into memory to make things generally faster. The user manual goes in depth about when to use it and you can read more about it here.
- ‘–fastq-input‘ specifies the reads file are in fastq format.
- ‘–output‘ specifies the output filename prefix. I generally call this ‘kraken.out.txt’
So first let’s grab the database:
cp -r /scratch/gencore/datasets/metagenomics/minikraken_20141208 ./
So the final command looks like this:
kraken --threads 4 \
--preload \
--fastq-input \
--output kraken.out.txt \
--db minikraken_20141208 \
kneaddata_output/SRR5813385.subset_kneaddata.trimmed.fastq
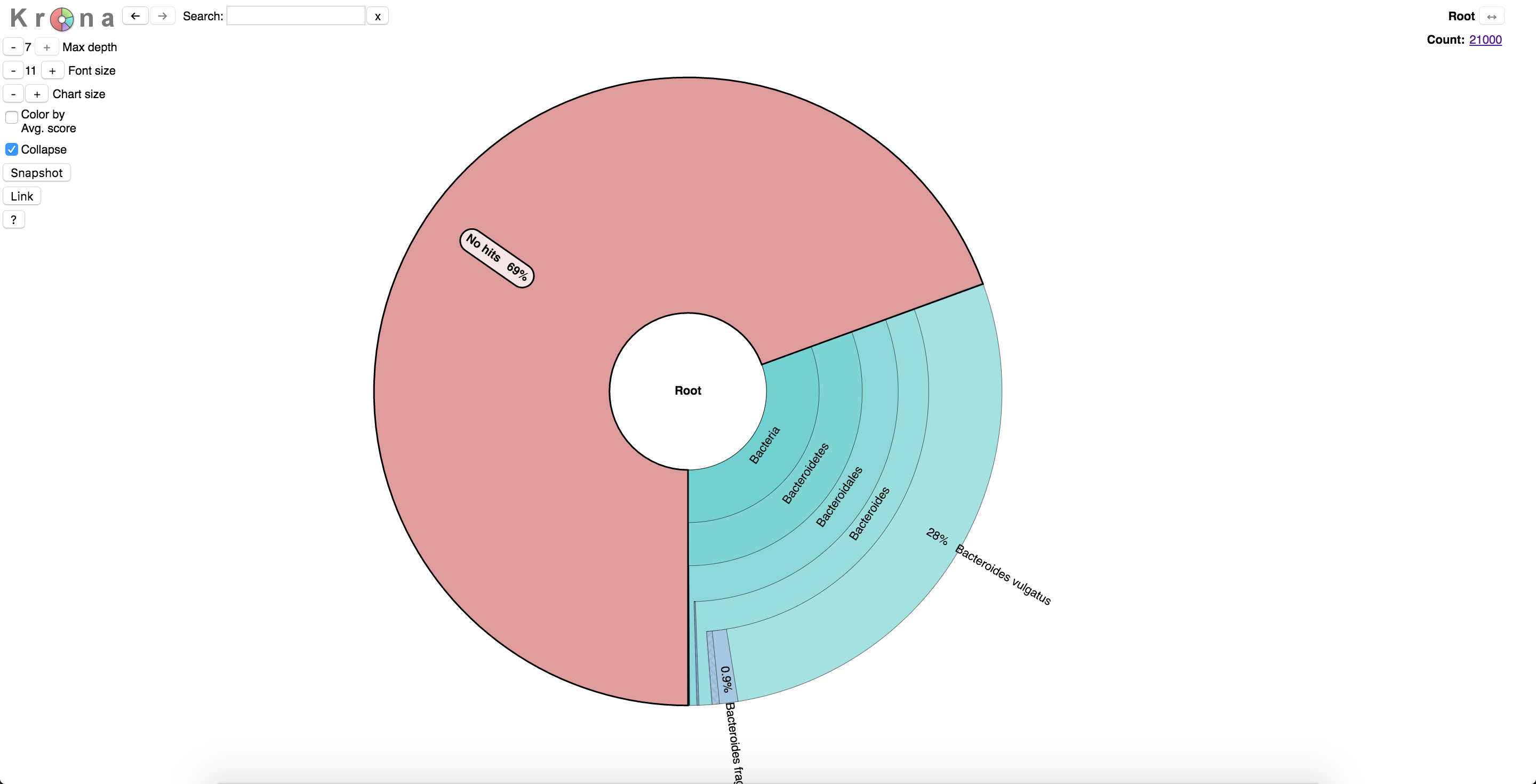
Looking at Kraken Results Using Krona
Now that Kraken has finished, we want to see what it found. To do this, we will use Krona to view the data. Krona allows hierarchical data to be explored with zoomable pie charts. Krona charts can be created using an Excel template or KronaTools, which includes support for several bioinformatics tools and raw data formats. The charts can be viewed with a recent version of any major web browser.
Krona requires an input file with two columns containing:
- A taxonomic identifier – this will be the NCBI taxonomic identifier
- A score – in this instance, it is the relative abundance of that taxonomic ID within your sample.
You can specify which columns hold the identifier and the score, using the ‘-t’ and ‘-s’ respectively. To run Krona using the Kraken output the command will look like this:
ktImportTaxonomy -o kraken.krona.html -t 3 -s 4 kraken.out.txt
The ‘-o’ parameter specifies the name of the output html file that you will view in a web browser.
Transfer the html file over to your computer and open it with a web browser.
You should see something like this. Note the 73% of unassigned reads – these can be either uninformative or unclassified. Downstream methods (specifically assembly) is very helpful in identifying the content. Remember, if your data doesn’t look right you can always rerun the filtering commands with different settings. Also, in non-targeted metagenomic sequencing it is normal to have a large number of unassigned reads.