scRNA-seq is a relatively new technology first introduced by Tang et al. in 2009, but the cost of sequencing and limited number of protocols at the time meant that it did not get widespread popularity until 2014.
scRNAseq differs from “traditional” bulk RNAseq. In bulk RNAseq, we measure the average expression of genes from multiple cells. This allows us to examine gene expression profiles between various conditions/treatments/timepoints etc, but is limiting when attempting to understand gene expression patterns within the cell. scRNA allows us to understand cellular differences in expression, and hence it is directly applicable to the studies of cell heterogeneity, cell population and subpopulation identification, effects of low copy mRNA distribution and transcriptional regulation. Perhaps more importantly, scRNAseq allows us to examine what would otherwise be considered as “noise”, in other words, the stochasticity of gene expression at the cellular level.
Below is a general overview of scRNA-seq
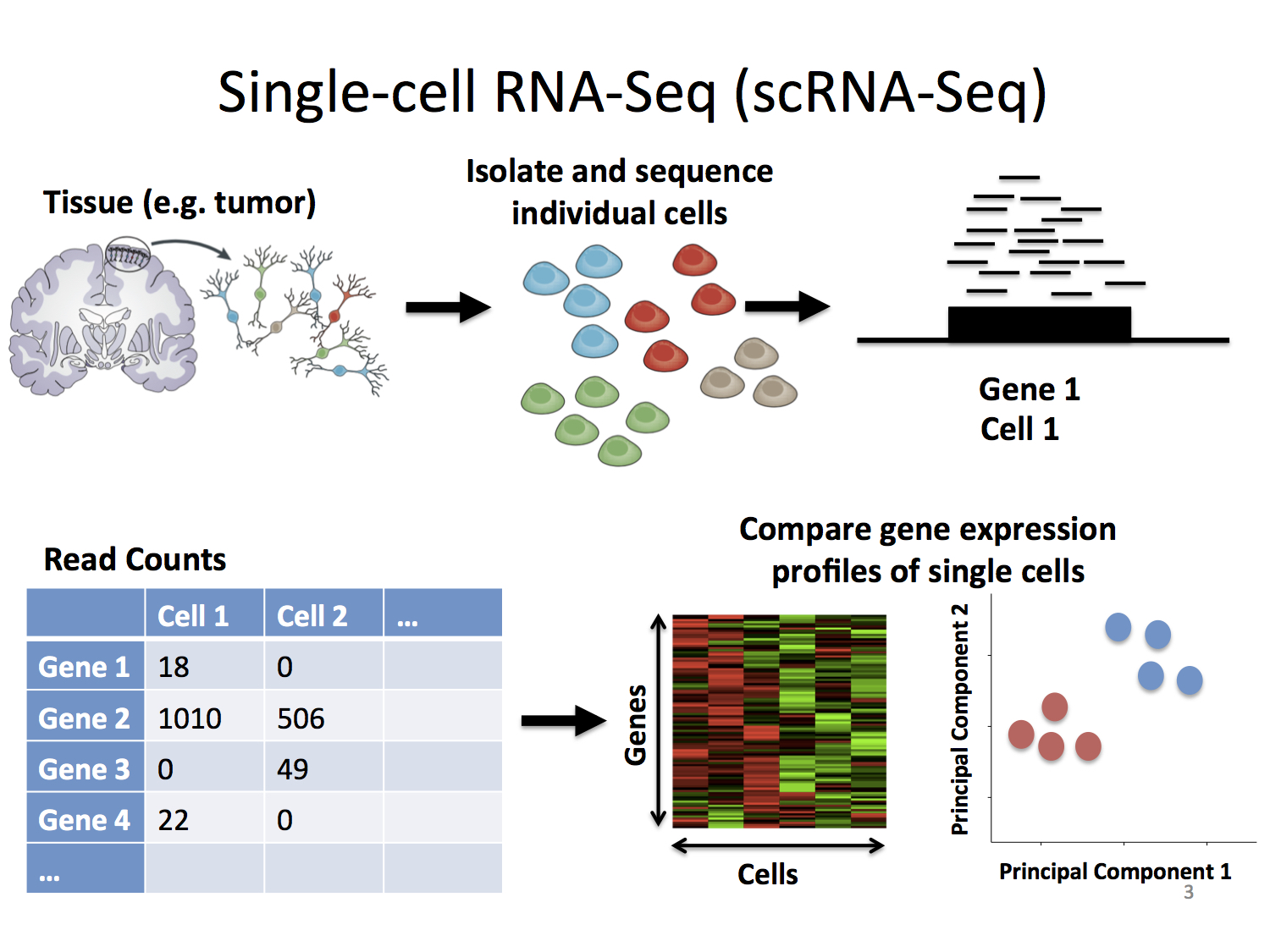
Stephanie Hicks
Welcome to the World of Single-Cell RNA-Sequencing
https://speakerdeck.com/stephaniehicks/welcome-to-the-world-of-single-cell-rna-sequencing?slide=3
There are still challenges that are associated with RNAseq analysis, such as experimental design, normalization as well as technical bias to name a few. In scRNA, some of these challenges are more prominent, for example dealing with low amounts of initial input leading to amplification biases, transcriptional “dropout” events as well as transcriptional “bursting” or “pulsing”.
Luckily, and since scRNAseq has gained popularity from ~2014 onwards, there are now multiple tools and published guidelines relating to the analysis of scRNA data. There are also multiple platforms and protocols available to choose from, according to the experimental design and biological questions being asked.
In this exercise, we will examine one popular tool tailored for scRNAseq analysis called Seurat. We will focus on the Seurat guided tutorial using 2,700 PBMC cells (publicly available from 10X genomics).The tutorial is available in full HERE.
Seurat is an R package designed for QC, analysis, and exploration of single cell RNA-seq data. Seurat aims to enable users to identify and interpret sources of heterogeneity from single cell transcriptomic measurements, and to integrate diverse types of single cell data (http://satijalab.org/seurat/).
We will also show how you could import your own gene expression matrix in Seurat and create a “seurat” object with it, since this seems to be a popular howto for R and scRNA beginners.
For an online R shiny app instance of Seurat (and other RNAseq analysis tools), have a look at our TSAR (Transcriptome Sequencing Analysis Resource) instance.