Required modules (NYUAD-Dalma)
The software modules at NYUAD’s HPC (Dalma) have been grouped according to analysis disciplines. For this tutorial, you will need the following modules.
module load gencore/1 module load gencore_variant_detection/1.0
Required software
Deeptools2 – deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq.
There are 3 ways for using deepTools:
- Galaxy usage – deepTools Galaxy server
- command line usage – The way we use deeptools at NYUAD, and it’s available through the gencore_variant_detection module.
- API – make use of your favorite deepTools modules in your own python programs (see deepTools API)
The image below (taken from the deeptools website), summarizes the funclionalities (and logic) of deeptools. And although we are using deeptools within the context of CHiP-seq analysis, it is very versatile and can be used for multiple analysis types.
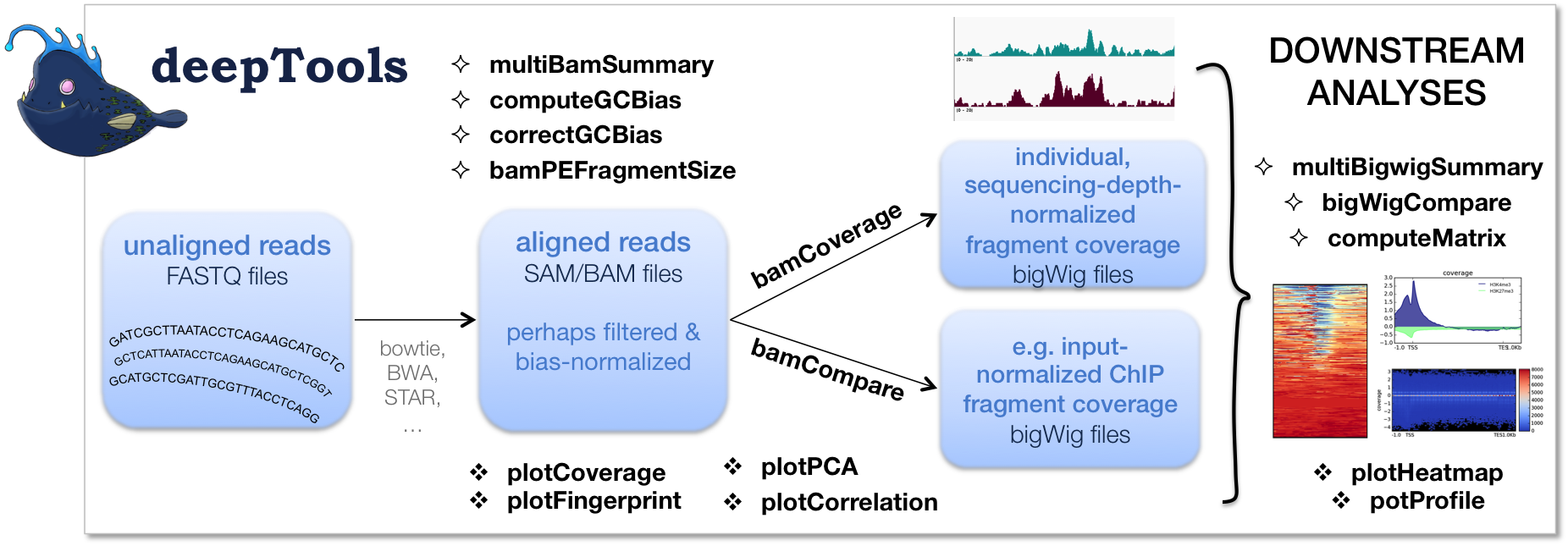
Introduction
ChiP-seq combines chromatin immunoprecipitation with high throughput sequencing and is used to analyze protein interaction with DNA and identify their binding sites. Understanding how proteins bind (e.g. looking at transcription factor binding and histone modifications) helps in understanding how genes (and ultimately phenotypes) are regulated and affected. ChipSeq falls under the epigenetic analysis category, and similar to other analysis categories, they involve a range of applications, tools and methods, which are all available to NYUAD HPC users by loading the appropriate module.
The image below outlines a typical ChipSeq preparation workflow,
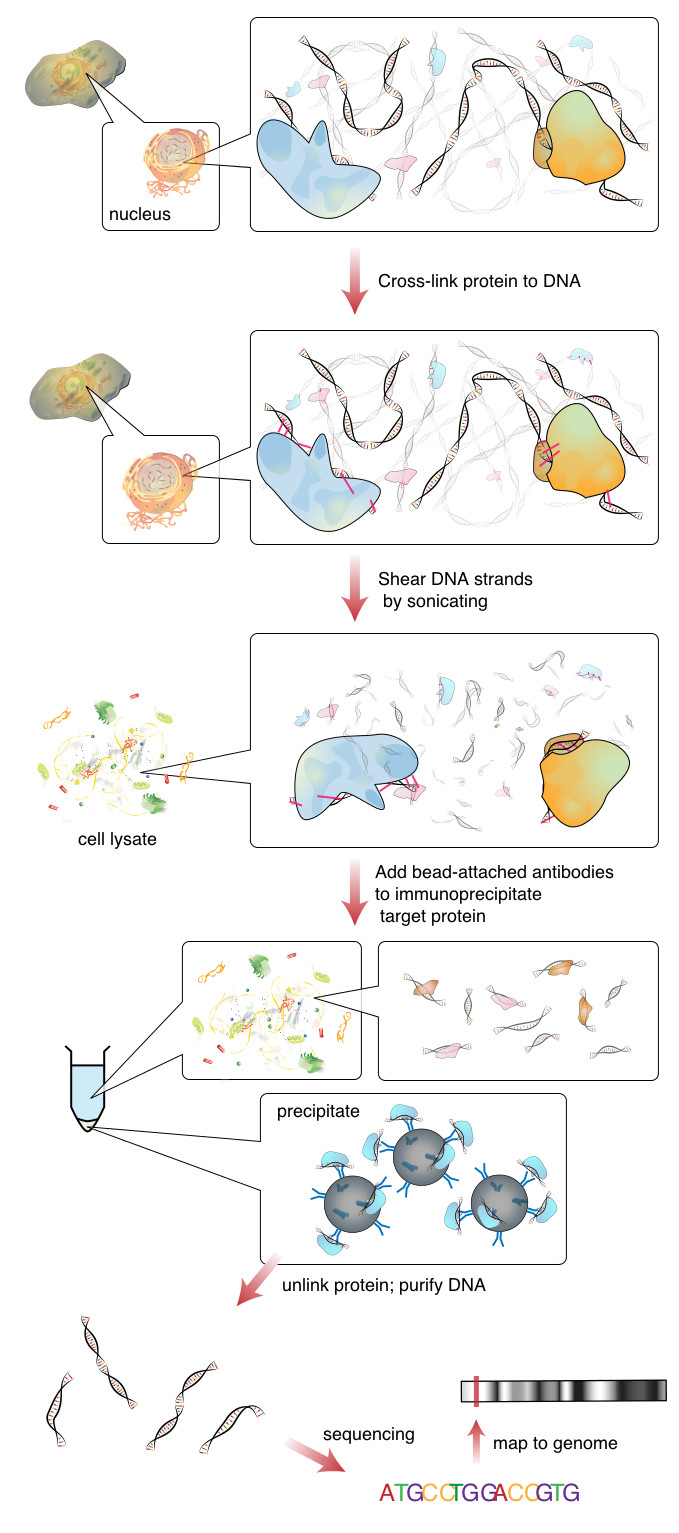
In this tutorial, we will retrace the bioinformatics analysis in (Xin et al) using deeptools. Although we won’t be calling peaks, deeptools provides some easy to use methods for assessing and analyzing CHiP-seq data. We will also be using the results of an RNA-seq differential gene expression analysis, in order to integrate (an overlay) these two genomics datasets. Ultimately, we are interested in understanding what epigenetic modifications are happening along the sets of DEGs that we have identified from the RNA-seq in WT and KO samples.