The official documentation for the GFF3 format can be found here
General Feature Format (GFF) is a tab-delimited text file that holds information any and every feature that can be applied to a nucleic acid or protein sequence. Everything from CDS, microRNAs, binding domains, ORFs, and more can be handled by this format. Unfortunately there have been many variations of the original GFF format and many have since become incompatible with each other. The latest accepted format (GFF3) has attempted to address many of the issues that were missing from previous versions.
GFF3 has 9 required fields, though not all are utilized (either blank or a default value of ‘.’).
- Sequence ID
- Source
- Describes the algorithm or the procedure that generated this feature. Typically Genescane or Genebank, respectively.
- Feature Type
- Describes what the feature is (mRNA, domain, exon, etc.).
- These terms are constrained to the [Sequence Ontology terms](http://www.sequenceontology.org/).
- Feature Start
- Feature End
- Score
- Typically E-values for sequence similarity and P-values for predictions.
- Strand
- Phase
- Indicates where the feature begins with reference to the reading frame. The phase is one of the integers 0, 1, or 2, indicating the number of bases that should be removed from the beginning of this feature to reach the first base of the next codon.
- Atributes
- A semicolon-separated list of tag-value pairs, providing additional information about each feature. Some of these tags are predefined, e.g. ID, Name, Alias, Parent . You can see the full list [here](https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md).
GFF3 Example
The canonical gene can be represented by the following figure
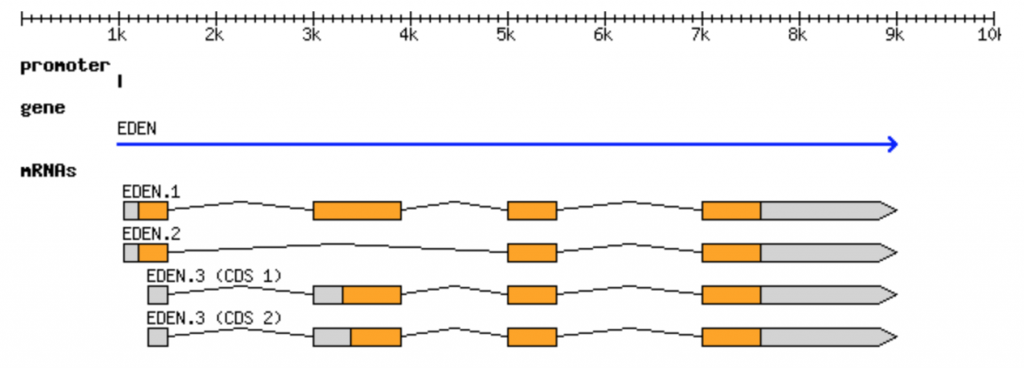
The same information can be represented in GFF3 format:
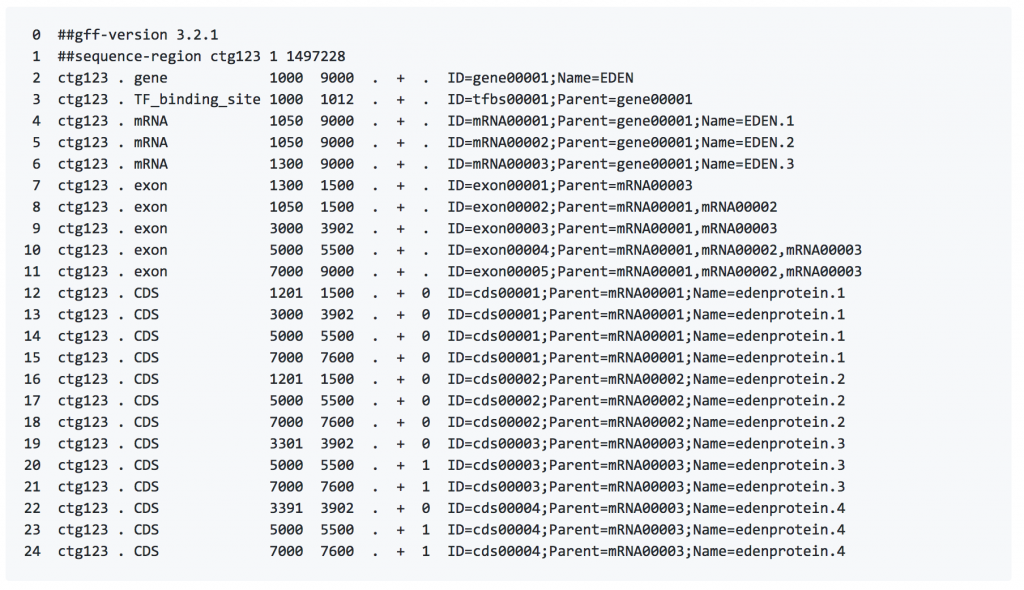
What Software uses GFF3?
- Any tool that requires information about gene position for analysis such as:
- Mapping RNA-seq such as [Tophat](https://ccb.jhu.edu/software/tophat/index.shtml), [HTSeq](https://htseq.readthedocs.io/en/release_0.9.1/)
- Genome Browsers like [IGV](http://software.broadinstitute.org/software/igv/), [Gbrowse](http://gmod.org/wiki/GBrowse), [UCSC](https://genome.ucsc.edu/)
How is this file generated?
- Feature identification software report motifs/features in this format.
- Almost all sequence annotation databases report in this format.
Let’s grab one!
Download it into your directory: ftp://ftp.wormbase.org/pub/wormbase/species/c_elegans/gff/c_elegans.WS236.annotations.gff3.gz
Take a look at it and see what it looks like!