The theory behind aligning RNA sequence data is essentially the same as discussed earlier in the book, with one caveat: RNA sequences do not contain introns. Gene models in Eukaryotes contain introns which are often spliced out during transcription. RNA Sequences that span two exons will have a hard time mapping to the genome, which still contains the code for introns.
One solution is to map the RNA sequence data to the predicted RNA molecules. However there are several disadvantages of mapping the RNA sequences directly to the predicted transcripts:
- We have to rely on the accuracy of the gene structure prediction method. Many genes structures are incomplete and also inaccurate.
- The genes we identify in the RNA samples may not be annotated in the genome yet. In this case, we would completely miss the gene from our analysis.
- Mapping the sequences to the genome can help us identify the genes that are missing from our annotation or annotated incorrectly.
Tophat
There are several softwares out now that have been developed specifically to help align RNA reads to the genome. One of the earliest and most popular is Tophat. Tophat is built on top of Bowtie ( another popular short read aligner aligned based on BWT ).
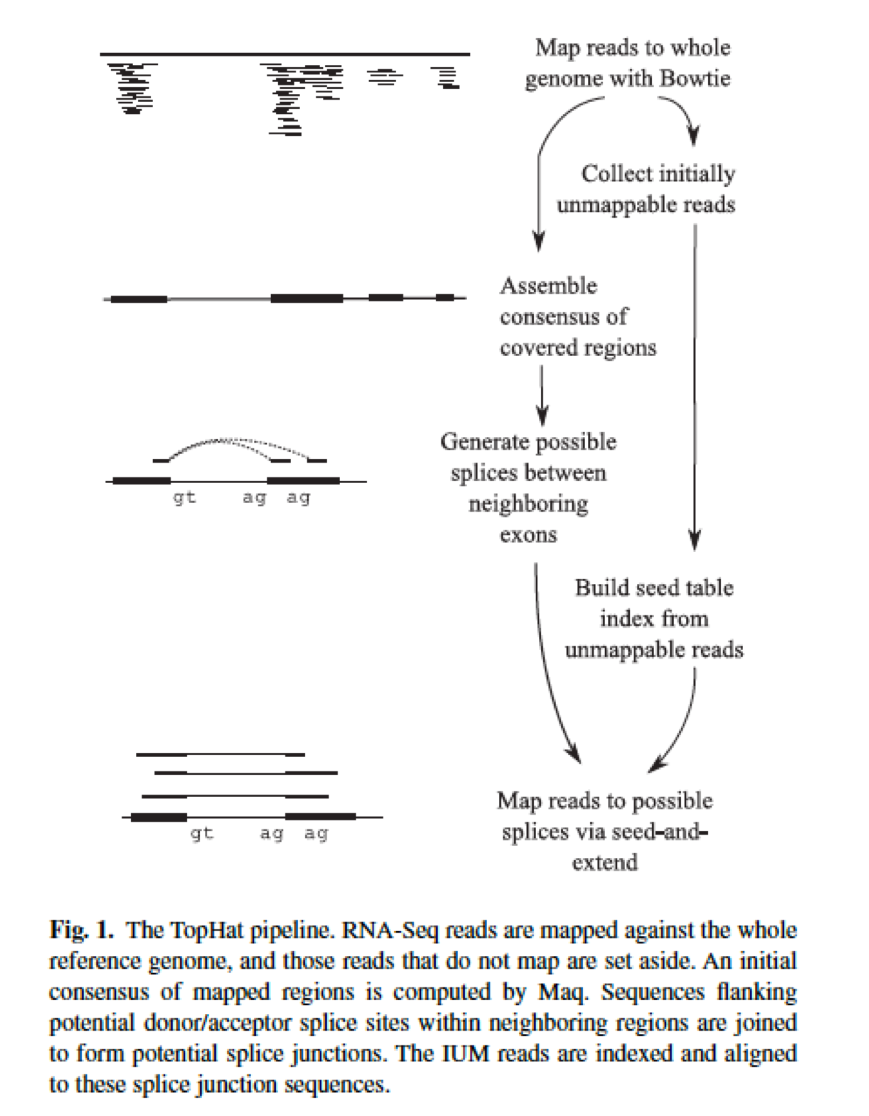
(Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111.)
Other RNA-seq aligners
There have been some newer RNA-seq aligners that are worth considering:
- HISAT: http://www.ccb.jhu.edu/software/hisat/index.shtml
- STAR: https://code.google.com/archive/p/rna-star/
- Kallisto: http://pachterlab.github.io/kallisto/manual.html
- Salmon: http://salmon.readthedocs.io/en/latest/salmon.html
Workflow using Tophat as an aligner
Let’s copy the data set into our scratch directory, unzip and untar the files.
$ cd $SCRATCH
$ cp /scratch/gencore/datasets/rnaseq_prealign.tar.gz .
$ gunzip rnaseq_prealign.tar.gz
$ tar xvf rnaseq_prealign.tar
rnaseq_prealign/
rnaseq_prealign/rnaseq.sh
rnaseq_prealign/Athaliana.fa
rnaseq_prealign/NO3_2.fastq.gz
rnaseq_prealign/trim.sh
rnaseq_prealign/tophat.sh
rnaseq_prealign/KCL_1.fastq.gz
rnaseq_prealign/Arabidopsis.gtf
rnaseq_prealign/bowtiebuild.sh
rnaseq_prealign/fastq.array.sh
rnaseq_prealign/KCL_2.fastq.gz
rnaseq_prealign/NO3_1.fastq.gz
rnaseq_prealign/dedup.sh
rnaseq_prealign/Athaliana.sorted.gff
$ cd rnaseq_prealign
$
Fastqc
First we will start with running the quality control for our sequences. We will use the software Fastqc, and to run this the command is very simple; just type fastqc and then the name of the file. Since there are only 4 files so we could simply run it 4 times, however let’s spend the extra time to create an array job which will look for all the fastq file in the directory and run fastqc. This will save us from significant amount of work in the future.
fastq.array.sh
#!/bin/bash
#SBATCH --array=0-3
#SBATCH --output=arrayJob_%A_%a.out
#SBATCH --error=arrayJob_%A_%a.err
#SBATCH -J fastqc
#SBATCH -p serial
# the below code finds all files that end with fastq.gz and saves it as an array called FILES
FILES=($(ls *fastq.gz))
# this is going to print out the name of the file -> it will be saved in our output
echo ${FILES[$SLURM_ARRAY_TASK_ID]}
# this is actually doing the work.
module load gencore/1
module load gencore_qc/1.0
fastqc ${FILES[$SLURM_ARRAY_TASK_ID]}
# I like to print this to the output so i know the script completed successfully.
echo "done"
# to execute script
sbatch fastq.array.sh
Fastqc provides an .html file that can be opened in a browser and a .zip file that contains the html file and other information in a parsable text file. You may want to transfer these files to your computer to view them. Our array SLURM job also produced one .err and one .out file for each job that was executed.
$ ls -l *zip *html *err *out
-rw-rw-r--. 1 mkatari mkatari 893 Jan 8 17:04 arrayJob_65642_0.err
-rw-rw-r--. 1 mkatari mkatari 57 Jan 8 17:04 arrayJob_65642_0.out
-rw-rw-r--. 1 mkatari mkatari 853 Jan 8 17:04 arrayJob_65642_1.err
-rw-rw-r--. 1 mkatari mkatari 57 Jan 8 17:04 arrayJob_65642_1.out
-rw-rw-r--. 1 mkatari mkatari 853 Jan 8 17:04 arrayJob_65642_2.err
-rw-rw-r--. 1 mkatari mkatari 57 Jan 8 17:04 arrayJob_65642_2.out
-rw-rw-r--. 1 mkatari mkatari 853 Jan 8 17:04 arrayJob_65642_3.err
-rw-rw-r--. 1 mkatari mkatari 57 Jan 8 17:04 arrayJob_65642_3.out
-rw-rw-r--. 1 mkatari mkatari 333669 Jan 8 17:04 KCL_1_fastqc.html
-rw-rw-r--. 1 mkatari mkatari 444271 Jan 8 17:04 KCL_1_fastqc.zip
-rw-rw-r--. 1 mkatari mkatari 338392 Jan 8 17:04 KCL_2_fastqc.html
-rw-rw-r--. 1 mkatari mkatari 449652 Jan 8 17:04 KCL_2_fastqc.zip
-rw-rw-r--. 1 mkatari mkatari 322399 Jan 8 17:04 NO3_1_fastqc.html
-rw-rw-r--. 1 mkatari mkatari 425541 Jan 8 17:04 NO3_1_fastqc.zip
-rw-rw-r--. 1 mkatari mkatari 341252 Jan 8 17:04 NO3_2_fastqc.html
-rw-rw-r--. 1 mkatari mkatari 453506 Jan 8 17:04 NO3_2_fastqc.zip
A quick glance at the reports show us that :
- The sequence quality starts to decrease as we get closer to the 3′ end of the read
- Something happened to the tile which caused the middle portion to have a significant lower quality
- There are duplicated sequences which tend to be the Illumina adapters
- And there is also an over represented K-mers in the 5′
- The quality encoding is using Illumina 1.5, so when we run an analysis that will use quality scores, make sure it is using the correct encoding (phred64). For details see (https://en.wikipedia.org/wiki/FASTQ_format)
Trimmomatic
Trimmomatic has options to:
- Remove leading and trailing nucleotide based on quality or simply a given number of bases
- Remove sequence when the average quality of a window falls below a certain threshold
- Remove sequences matching Illumina adapters.
To run Trimmomatic, we can use the same strategy as above.
trim.sh
#!/bin/bash
#SBATCH --array=0-3
#SBATCH --output=Trimmomatic_%A_%a.out
#SBATCH --error=Trimmomatic_%A_%a.err
#SBATCH -J Trimmomatic
#SBACTH -p serial
# the below code finds all files that end with fastq.gz and saves it as an array called FILES
FILES=($(ls *fastq.gz))
# this is going to assign the variables to file names
INPUT=${FILES[$SLURM_ARRAY_TASK_ID]}
OUTPUT=${INPUT}.trimmed.fastq
# this is actually doing the work.
module load trimmomatic
# make sure we use phred64
trimmomatic SE -phred64 \
$INPUT \
$OUTPUT \
HEADCROP:10 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
# I like to print this to the output so i know the script completed successfully.
echo "done"
# to execute script
sbatch trim.sh
Tophat
In order to align your RNA sequences to the genome with Tophat, you have to first create the database files using bowtie. bowtie2-build needs the fasta file as the first argument followed by the prefix to be used for the database index files.
bowtiebuild.sh
#!/bin/bash
#SBATCH -J BowtieBuild
#SBATCH -p serial
#SBATCH --output=BowtieBuild.out
#SBATCH --error=BowtieBuild.err
module load samtools
# indexing the fasta files comes in handy for later use but is not necessary for bowtie-build.
samtools faidx Athaliana.fa
module load bowtie2
# bowtie2-build bowtie2-build Athaliana.fa Athaliana
# to execute script
sbatch bowtiebuild.sh
Then we can take each sample and align them against the Arabidopsis genome.
tophat.sh
#!/bin/bash
#SBATCH --array=0-3
#SBATCH --output=Tophat_%A_%a.out
#SBATCH --error=Tophat_%A_%a.err
#SBATCH -J Tophat
#SBATCH -n 4
#SBATCH --time=01:00:00
#SBATCH -p serial
module purge
module load tophat2
FILES=($(ls *trimmed.fastq))
# this is going to assign the variables to file names
INPUT=${FILES[$SLURM_ARRAY_TASK_ID]}
OUTPUT=${INPUT}.tophat
tophat2 -i 10 -p 4 --no-coverage-search \
--solexa1.3-quals \
-o $OUTPUT \
-G Arabidopsis.gtf \
Athaliana \
$INPUT
# --solexa1.3-quals specifying that quality score is phred64
# -i = the minimum inton size
# -o = the directory where the output should be saved.
# Here I have used a variable that can be assigned in the bash script
# -G = a genome annotation to use as a reference.
# The last two parameters are the prefix to the Bowtie database
# and then finally the fastq file containing the RNA sequence data.
# This is also a variable that you can assign.
# to execute script
sbatch tophat.sh
You want to run this command separately for all the samples. The option to align paired reads is slightly different.
Now we can sort and de-duplicate the reads. Sorting and indexing are common transformations that allow applications to process the data efficiently. Picard has an excellent collection of tools that can be used. There is also samtools, which has some equivalent functions, however I find that I run into fewer errors when I use Picard. (https://broadinstitute.github.io/picard/)
dedup.sh
#!/bin/bash
#SBATCH --array=0-3
#SBATCH --output=Dedup_%A_%a.out
#SBATCH --error=Dedup_%A_%a.err
#SBATCH -J Dedup
#SBATCH --time=01:00:00
#SBATCH -p serial
# get all tophat directories
DIRS=($(ls -d *tophat))
# names are getting too long so let's cut out and grab the important part of the name
INPUTNAME=($(echo ${DIRS[$SLURM_ARRAY_TASK_ID]} | cut -d '.' -f 1))
module load picard
# sort the bam file. All output files are called accepted_hits.bam in the tophat
# output directory.
picard SortSam \
INPUT=${DIRS[$SLURM_ARRAY_TASK_ID]}/accepted_hits.bam \
OUTPUT=${INPUTNAME}_sorted.bam \
SORT_ORDER=coordinate
# remove duplications
picard MarkDuplicates \
INPUT=${INPUTNAME}_sorted.bam \
OUTPUT=${INPUTNAME}_dedup.bam \
METRICS_FILE=${INPUTNAME}.dedup.metrics \
REMOVE_DUPLICATES=TRUE \
ASSUME_SORTED=TRUE \
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000
# create index files for the bam files. This is helpful if you want to visualize your results.
picard BuildBamIndex \
INPUT=${INPUTNAME}_dedup.bam \
OUTPUT=${INPUTNAME}_dedup.bam.bai
echo "dedup done"
# to execute script
sbatch dedup.sh
Now that we have the alignment files processed, we can use one many different tools to determine differentially expressed genes.