In any high-throughput sequencing experiment there are pieces of information that we are not interested in and data that retains no value. Be it low quality, ambiguous bases, or contaminating sequences, no matter the application, there will always be some noise. In particular, when obtaining samples from an environment, there are many factors that can add to the level of noise when it comes time to sequence.
The goal of metagenomics is to sequence everything in your sample, hence the prefix “meta.” Unfortunately the sample collection methods, as well as the library prep protocols, are not capable of completely omitting contaminating sequences. Obtaining metagenomic samples from a host (human, bees, sponges, etc.), a fairly common application of metagenomics, will result in host DNA/RNA in your sequencing data, and generally at large quantities. With that said, it is of utmost importance to remove the confounding data to help purify an already complex dataset. This starts with removing low quality sequences/bases as well as sequences that you believe to be present that you are not interested in.
Quality Filtering and Trimming
In any analysis, this should be the first step. Not only will this give you an idea of the quality of your data but it will also clean up and reduce the size of your data, making downstream analysis much easier! The main steps in this process are:
- Removing low quality bases
- Removing low complexity reads
- Remove artifacts (barcodes, adapters, chimeras)
To do this, you can review the trimming tutorial discussed earlier in this workshop.
Contaminant Removal
This step is somewhat unique to metagenomic analyses as the prep methods are non discriminant and will sequence everything that is in your sample, including all contaminants. When it comes to assembly and taxonomic calculations this can really complicate the results as well as add to compute time.
For every analysis you should always consider some potential contaminants and subsequently gather the necessary reference sequences:
- Was PhiX added to your sequencing run?
- If yes, this will always be present in your sample (though generally at lower concentrations).
- The [PhiX genome](https://www.ncbi.nlm.nih.gov/nuccore/9626372) can be found here.
- Did your samples come from a host?
- If yes, you’ll want to grab the genomes for these to remove them.
- Does your lab work with any other organisms’ DNA?
- If yes, it is possible that some of its DNA has contaminated your sample.
- Is this a meta-RNA-Seq analysis?
- If yes, you will want to remove/normalize rRNA genes.
- You can use an [rRNA database](https://www.arb-silva.de/) or other rRNA removal software, such as [SortMeRNA](http://bioinfo.lifl.fr/RNA/sortmerna/).
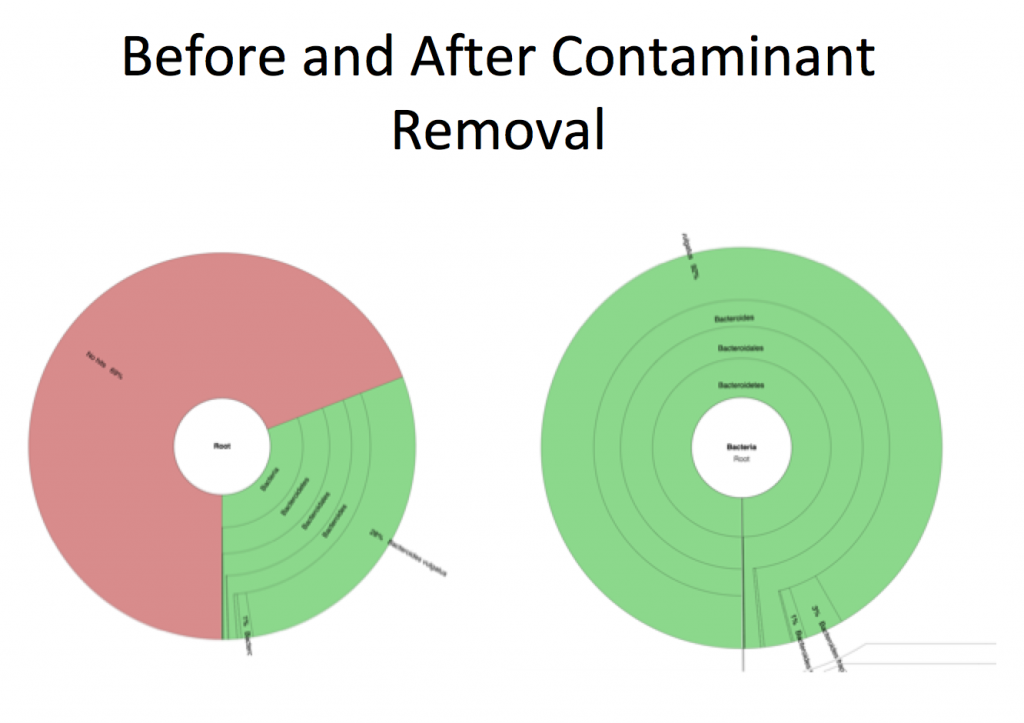
How to Remove Contaminants
The first step is to gather your reference sequences of the organisms/sequences you do not want in your sample. Once you have all your contaminant sequences, put them all in a one file and index it with bowtie2.
Note: Indexing with bowtie2 is only a requirement for the KneadData software.
To create this reference database, you can use the cat command and the
>
and
>>
redirects to write and append sequences to a preexisting reference file, respectively.
To add all sequences to a file you can type:
cat file1.fasta file2.fasta file3.fasta > references.fasta
To append files to an existing genome file:
cat file1.fasta file2.fasta >> references.fasta
Then when you have all of your sequences in a file, you can index it with bowtie2:
bowtie2-build references.fasta references
For this tutorial, we will only use the human genome. You can download it with KneadData using the following command, though I have already provided this:
kneaddata_database --download human_genome bowtie2 ./
This will download the bowtie2 indexed human genome into the current directory.
NOTE: For targeted queries, it is not always necessary to remove contaminant sequences as they are generally not represented in the databases. However, there are some exceptions and it is good practice to always filter your data.
KneadData
KneadData invokes Trimmomatic for its quality filtering/trimming, as well as Tandem Repeat Finder (TRF) and FastQC, and Bowtie2 to align your reads to your list of contaminant reference sequences. In essence, Trimmomatic is capable of throwing away reads or parts of reads that have low quality scores, as well as trimming adaptor sequences. TRF finds tandem repeats and removes them, while FastQC generates a quality report for your data. Bowtie2 will throw out anything that aligns to the sequences that you don’t want to include in your analysis. These are common problems that arise in almost all sequencing runs and should be handled appropriately.
To run KneadData you need:
- An indexed contaminant database
- Reads in fastq format
To get started, enter an interactive session:
srun --mem 15GB --cpus-per-task 4 --time 3:00:00 --pty /bin/bash
module purge all
module load gencore/1 gencore_metagenomics
Create a directory called metagenomics and transfer over the files that I have prepared for you:
cd $SCRATCH
mkdir metagenomics
cd metagenomics
cp /scratch/gencore/datasets/metagenomics/Homo_sapiens_Bowtie2_v0.1/Homo_sapiens.* ./
cp /scratch/gencore/datasets/metagenomics/SRR5813385.subset.fastq ./
Now we want to filter our data using KneadData. This can be done as follows:
kneaddata --input SRR5813385.subset.fastq \
--bowtie2-options "--very-sensitive -p 4" \
--trimmomatic "${EBROOTGENCORE_METAGENOMICS}/share/trimmomatic-0.36-3" \
--reference-db Homo_sapiens \
--output kneaddata_output
This will create files in the folder namedkneaddata_output
SRR5813385.subset_kneaddata_Homo_sapiens_bowtie2_contam.fastq: FASTQ file containing reads that were identified as contaminants from the database.SRR5813385.subset_kneaddata.fastq: This file includes reads that were not in the reference database.SRR5813385.subset_kneaddata.trimmed.fastq: This file has trimmed reads.SRR5813385.subset_kneaddata.log: output from kneaddata
Now your data is much better than it was at the beginning and you can proceed to the next step. However, upon inspection of your data post filtering, it may be necessary to remove more contaminants and/or toggle the quality filtering parameters.