The official SAM documentation can be found here.
These formats were introduced to standardize how alignments are reported. Initially there were many different formats, most of them proprietary, which were space inefficient and either held too much or too little information. The first of these to be introduced was Sequence Alignment Map (SAM). With this format not only is the alignment retained but the associated quality scores (both mapping and base quality), the original read itself, paired-end information, sample information, and many more features.
SAM Format
This is the most basic, human readable format of the three. This is generated by almost every alignment algorithm that exists. It consists of a header, a row for every read in your dataset, and 11 tab-delimited fields describing that read.
SAM Header
The header varies in size but adheres to a particular format depending on what information you decide to add. Some example information that can be entered into the header is: command that generated the SAM file, SAM format version, sequencer name and version. The full list of available header fields can be found below.
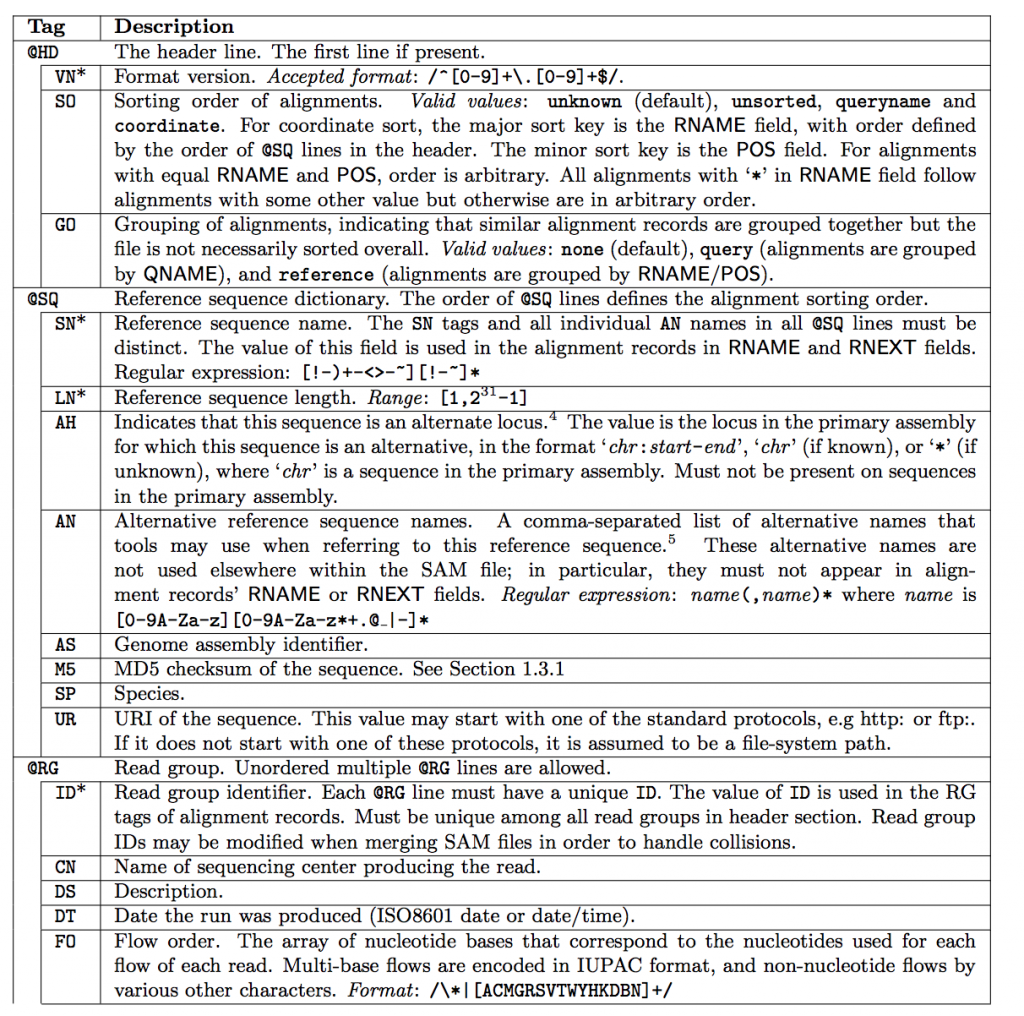
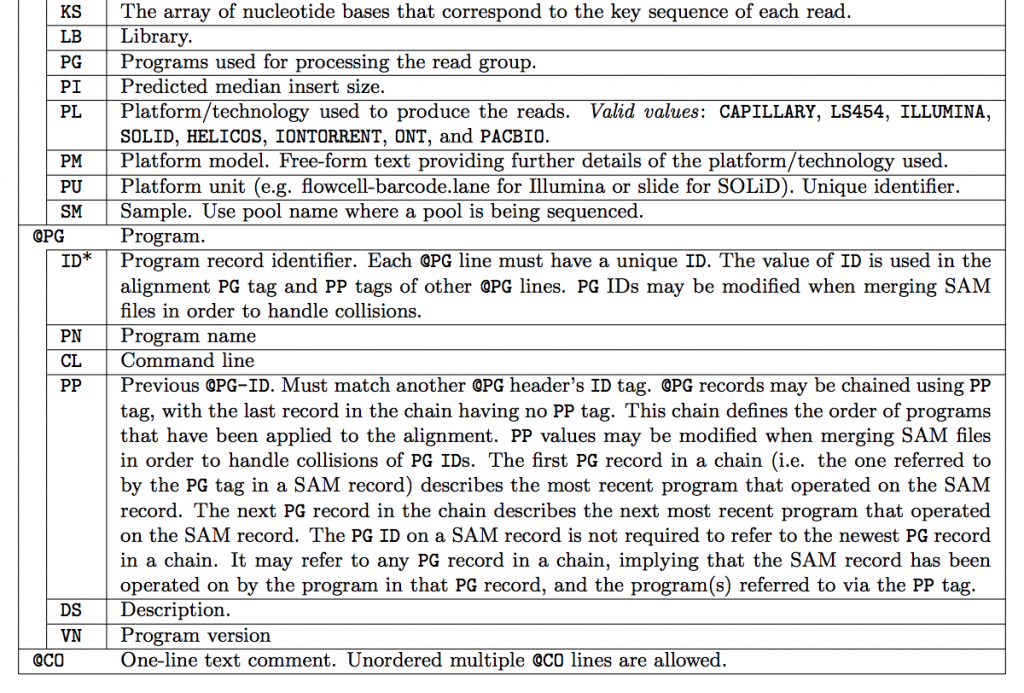
Field Descriptions
Each row contains 11 mandatory fields. The descriptions for them can be found below:
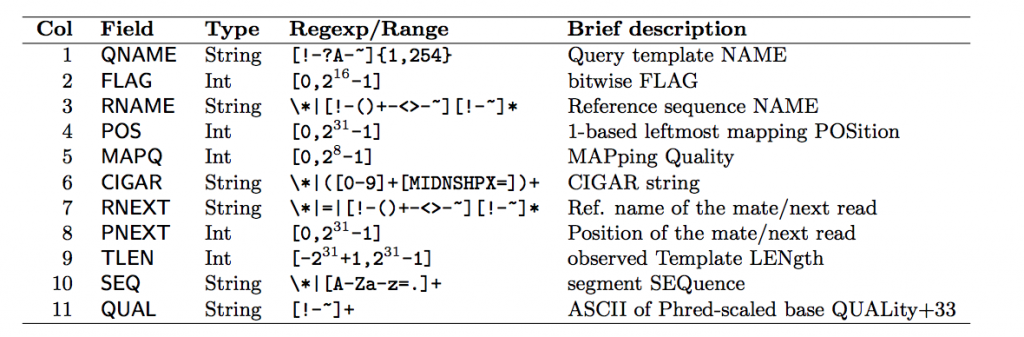
Let’s look at some of the fields that aren’t very self explanatory:
Bitwise Flag
The bitwise flag is a lookup code to explain certain features about the particular read (exact same concept as Linux permission codes!). It tells you whether the read aligned, is marked a PCR duplicate, if it’s mate aligned, etc. and any combination of the available tags, seen below:

One important thing to note is that any combination of these flags results in one integer, which makes interpreting it a bit difficult. To make it easy you can check here to either encode or decode a bitwise flag.
MapQ (Mapping Quality)
This value reports how well the read aligned to the reference. Different algorithms report it differently but nonetheless, the greater the number the better the alignment (generally).
CIGAR String
This is a shorthand way to encode an entire alignment. Instead of writing the whole alignment out, operators have been defined and are used in combination with numbers to explain which part of the sequence aligns, which doesn’t, and everything in between. The definition for the operators can be found here:

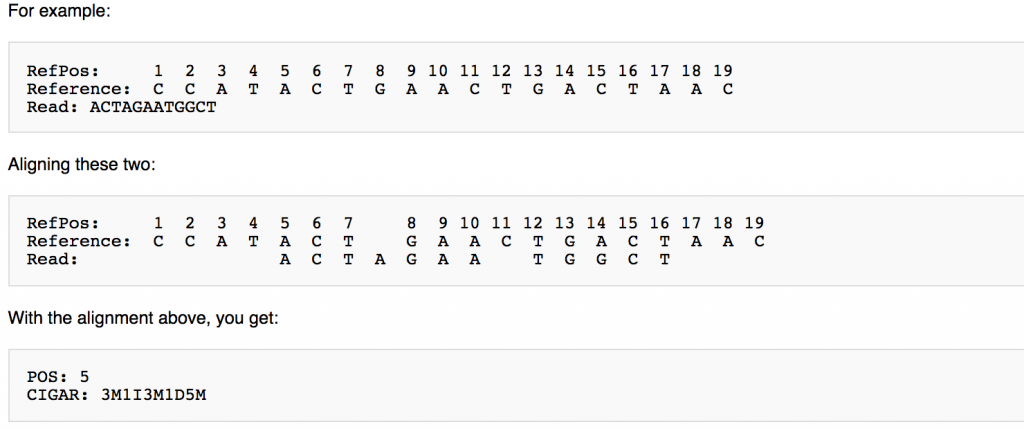
Example SAM file
HWI-ST865:416:C6CG0ACXX:1:1313:9073:43827 0 I 2 1 99M * 0 0 CCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT @C@DFDEFFHDFFIIJIGIIGIGGIIIIJGHGIJJEEIAHHGGIGFH@HGCFGGGJJIIGDAFG@DGIHHHHHFFBB@CACEC6;?CDD?CDCAD>>AA AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:99 YT:Z:UU
HWI-ST865:416:C6CG0ACXX:1:1215:16359:6484 16 I 9 1 85M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCC EEEEFFFFDAGHHHIJJIIJJJJJIJJJJIJIJJIGIIJJJJJJJIJJJJJJJIIJJJIIJGJJIJJJJJJJHFHHHFFFFFCCB AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:85 YT:Z:UU
HWI-ST865:416:C6CG0ACXX:1:1113:14118:89232 16 I 15 1 100M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAA CAC@A>C@AADCAC@ACEEC@@BD?E;@CEHGCEIGIHAFHGGF;FCHBHFBHIGIIIJJJJJJJJJJJJJJJJJJJJJIJJJJJJJHHHHHFFFFFCCC AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU
HWI-ST865:412:C6CLLACXX:1:2315:12173:84819 16 I 49 1 70M * 0 0 GCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT @7)3CC=)CA;EBC>DAEDBDCDDDCDD@B?<DEDE399CBC<+>EAE<BDCEAE3DADDD<ABDD;1?? AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:70 YT:Z:UU HWI-ST865:412:C6CLLACXX:1:1201:19323:33842 16 I 71 0 100M * 0 0 AAGCCTAAGCCTAAGCCTAAGCCTAAGCCAAATCCCAAGCCTAAGCCTAAGCCTAAGCCTAAGCCAGAGCCTAAGCCTAAGCCTTAGCCTAAGCCTGATC DDDCCDCCACCCDCCCC>CAA@D@;BDHA3A7(@5/IIHFIIGEIIIIHEIIIIIGGIGIIIIIFDCIIIIIIIIGGIIGIHFFEIHDDBHFFDDDB@@@ AS:i:-33 XS:i:-33 XN:i:0 XM:i:8 XO:i:0 XG:i:0 NM:i:8 MD:Z:29T2G2T29T0A17A11A1G1 YT:Z:UU
HWI-ST865:412:C6CLLACXX:1:1215:19021:78287 16 I 93 1 66M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGC @=87=3GF=.:CFE;D@B3?3?BD9BC<>CJJJJJJJJJJJJJJJJIJJJJJIHHHHHFFEDAC@B AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:66 YT:Z:UU
BAM Format
This is the same format except that it encoded in binary which means that it is significantly smaller than the SAM files and significantly faster to read, though it is not human legible and needs to be converted to another format (i.e. SAM) in order to make sense to us.
Some special tools are needed in order to make sense of BAM, such as Samtools, Picard Tools, and IGV which will be discussed in some of the latter sections.
CRAM Format
This is a relatively new format that is very similar to BAM as it also retains the same information as SAM and is compressed, but it is much smarter in the way that it stores the information. It’s very interesting and up and coming but is a bit beyond the scope of this course. However, if you’re up for it you can read about it here.
What software use these files?
- Alignment algorithms
- Some assemblers
- CRAM/unaligned Bam (uBAM) can be a source of data delivery in some institutions: this cuts down significantly on storage space and transfer speed.
- Alignment viewers
- Variant detection algorithms
How are these files generated?
- This is output from aligners and assemblers
- This can also be used to deliver raw data
Let’s look at one!
The default tool for interacting with these formats is samtools.
Log into a compute node and run the following commands:
module purge
module load samtools/intel/1.6
cp /scratch/courses/HITS-2018/file_formats/208_1_merged_trimmomatic_trimmed_bt2.bam ./c_elegans.bam
samtools view -H c_elegans.bam
What does this information mean? Look at the size of the file. What tool do you use to view it?