Now that we have our BigWigs, we will use the command computeMatrix. This tool calculates scores per genome regions and prepares an intermediate file that can be used with plotHeatmap and plotProfiles. Typically, the genome regions are genes, but any other region defined in a BED file can be used. computeMatrix accepts multiple score files (bigWig format) and multiple regions files (BED format). This tool can also be used to filter and sort regions according to their score.
computeMatrix will take the coordinates of interest that we have from a BED file representing the TSS of the genes of interest (1 file for up regulated genes and 1 file for down regulated genes). We used the UCSC Table Browser to convert the genes of interest into BED format for input in computeMatrix.
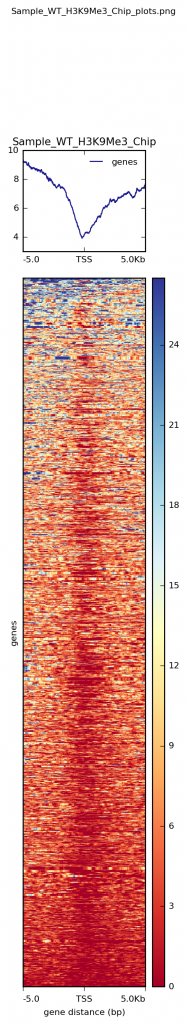
The output of computeMatrix will then be used in the command plotHeatmap to generate the heatmaps of the selected regions (genes in the BED files), similar to the image below.
The exercise
Since we are going to be using the NYUAD bioinformatics workflow system (BioSAILs) to submit our analysis jobs, the very first thing is to open the deeptools2_workflow.yml template and edit it. Let’s open it in Atom.
- Start Atom and open the deeptools2_workflow.yml.
Without going into too much detail regarding the “Global parameters” of the workflow, we are going to focus on the software section or “rules” section.
- Go to your terminal on Dalma and (in case you haven’t loaded the gencore and gencore_variant_detection modules load them now) and enter the following,
module load gencore module load gencore_variant_detection computeMatrix -h
(NOTE: Make sure you are at “/scratch/yournetid/chipseq_analysis/”)
This should now display the help menu for the command. Your task is to structure the computeMatrix command using the appropriate parameters. Here’s some information to guide your decisions,
- We are looking to generate a matrix using a reference-point approach.
- We want to scan upto 5kb either side of the TSS.
- We also want to add all the appropriate labels to our files.
Below is the code (in Atom) that we focusing on,
- deeptools2_computeMatrix:
local:
- outdir: '{$self->deeptools2_dir}/computeMatrix'
- INPUT: '{$self->inputdir}/{$sample}.bw'
- OUTPUT: '{$self->deeptools2_dir}/computeMatrix/{$sample}_computeMatrix.cp'
- HPC:
# - deps: 'deeptools2_bamCoverage'
- cpus_per_task: 12
- walltime: '08:00:00'
- mem: '50GB'
- module: 'gencore gencore_variant_detection'
process: |-
#TASK tags={$sample}
computeMatrix reference-point
-?? (input?) {$self->INPUT}
-?? (output?) {$self->OUTPUT}
-?? (downstreamOfTSS?) 5000 -?? (upStreamOfTSS?) 5000
--??(label?) {$sample}
-?? (threads?) 12
-?? (region file?) upReg.bed
We also need to do the same to plotHeatmap, so once again edit the plotHeatmap rule in the workflow and add the appropriate parameters. You can type “plotHeatmp -h” on the command line terminal to get an idea of the different parameters do.
- deeptools2_plotHeatmap:
local:
- outdir: '{$self->deeptools2_dir}/plotHeatmap'
- INPUT: '{$self->deeptools2_dir}/computeMatrix/{$sample}_computeMatrix.cp'
- OUTPUT: '{$self->deeptools2_dir}/plotHeatmap/{$sample}_plotHeatmap.png'
- HPC:
- deps: 'deeptools2_computeMatrix'
- walltime: '08:00:00'
- mem: '50GB'
- module: 'gencore gencore_variant_detection'
process: |-
#TASK tags={$sample}
plotHeatmap
-?? (input?) {$self->INPUT}
-?? (output?) {$self->OUTPUT}
--?? (label?) {$sample}
-?? (title?) {$sample}_plots.png
--?? (png format?) png
Finally, it’s time to execute the workflow. BioSAILs mainly revolves around 2 main commands, biox and hpcrunner.pl. biox will take as input a yml workflow template and generate an executable shell script, which can be submitted to an HPC scheduler using hpcrunner.pl. So,
- Make sure you are in the “chipseq_analysis” directory.
-
module purge all
- Load the BioSAILs modules
module load gencore module load gencore_dev
- Now we will use the biox command to create a shell script based on the workflow template. We need to supply the input workflow name, and the output shell script name,
biox run -w deeptools2_workflow.yml -o dt2.sh
- Finally, we can use the hpcrunner.pl command to submit the shell script that we created in the earlier (using biox) to the HPC (dalma) scheduler (SLURM). We also give a name for our project (chipseq_dt2),
hpcrunner.pl submit_jobs --infile dt2.sh --project chipseq_dt2
- Type the following command
squeue -u yournetid
to check on your jobs.
Copy data to your local machine
Once your jobs have completed (it should take between 10-15 minutes), you will need to copy over the the PNG images for each sample. These can be found in the “plotHeatmap” directories.
Also, choose (up to you) any 2 bigWig files and copy them over to your local machine as well (we will need them for the next section).