This step has already been performed for you. The reason is that it will not be feasible to transform the BAM alignment files to their representative BigWig files in the limited time that we have.
However, the section below highlights how this process works.
The bamCoverage command (part of the deeptools2 package) allows you to convert alignment files (in BAM format) to other alignment formats.
This tool takes an alignment of reads or fragments as input (BAM file) and generates a coverage track (bigWig or bedGraph) as output. The coverage is calculated as the number of reads per bin, where bins are short consecutive counting windows of a defined size. It is possible to extended the length of the reads to better reflect the actual fragment length. bamCoverage offers normalization by scaling factor, Reads Per Kilobase per Million mapped reads (RPKM), and 1x depth (reads per genome coverage, RPGC).
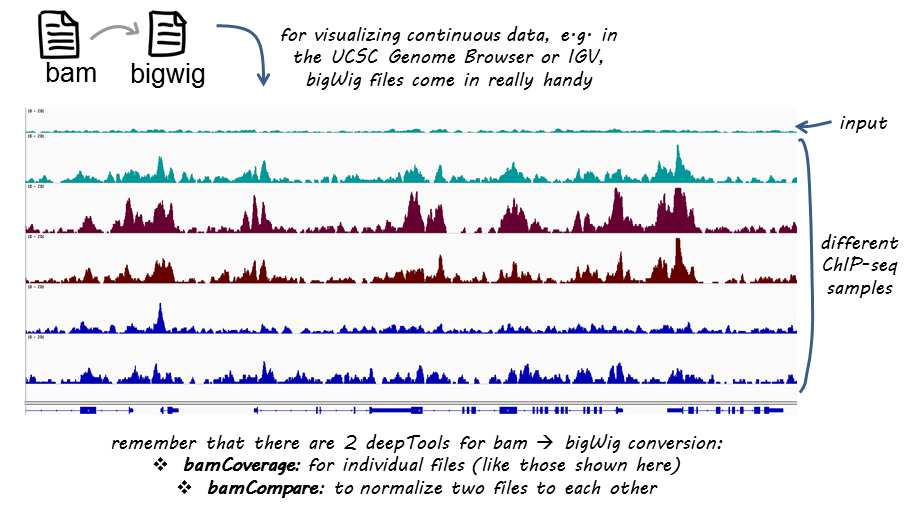
BigWig files have a much smaller data footprint compared to BAM files, especially as your bin size increases. It also allows for normalization, which is great if we want to compare different samples to each other (that vary in terms of sequencing depth).
In this case, for each one of our sample BAM alignments, we performed the following,
bamCoverage \ –of bigwig (output format)\ –bs 10 (bin size)\ -e (extend PE to frag size)\ --ignoreDuplicates \ --normalizeUsingRPKM \ -b (input bam)\ -o (output)\ -p 24
In this example, we chose to normalize the reads using RPKM (reads per million), and defining the bin size at every 10bp. The reason is that we want to maintain a high enough resolution. As such, our bigwigs are still a bit large, if we were to choose a bin size of 1,000bp or 10,000bp, the bigwigs would be much smaller. We also set the flag “–ignoreDuplicates”, and the reason for this is because we have processed our our aligned BAMs (using picard MarkDuplicates) in such a way as to remove duplicates rather than just mark them.
Note: In the “deeptools2_workflow.yml” file, the command above is commented out (lines starting with ‘#’), which means that it will be ignored. So if you wanted to run bamCoverage on your data (BAM alignments), all you have to do is edit the workflow file (deeptools2_workflow.yml) and delete the hashes ‘#’.