Hands On
The figure below depicts the simple pipeline we will build. Note the dependencies (indicated with arrows) and the steps which run in parallel (same row).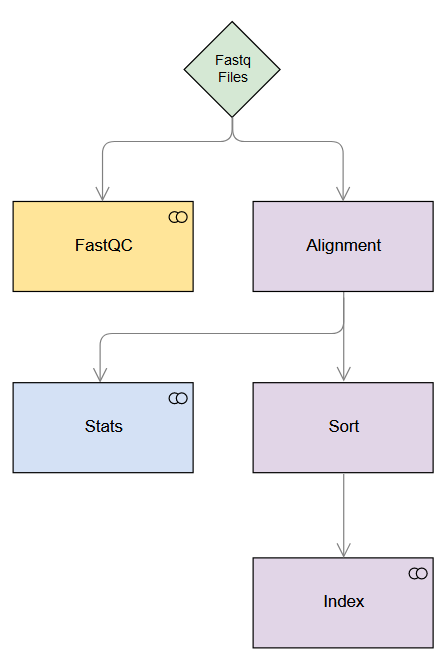
srun -c2 -t1:00:00 --mem=10000 --pty /bin/bashLoad nextflow
module load nextflow/20.01.0Make the config file
params.reads = "/scratch/mk5636/nextflow-badas/data/*.fastq.gz"
params.ref = "/scratch/work/cgsb/genomes/Public/Fungi/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa"
params.out = "$SCRATCH/nextflow-badas/out"
workDir = params.out + '/nextflow_work_dir'Building the workflow (
main.nf)
// Setup the reference file
ref = file(params.ref)
// Custom filenames
Channel
.fromFilePairs( params.reads, size: -1)
{ file -> file.getBaseName() - ~/_n0[12]/ - ~/.fastq/ }
.ifEmpty { error "No reads matching: ${params.reads}" }
.set { read_pairs_ch }
// Standard Illumina filenames
//Channel
// .fromFilePairs( params.reads )
// .ifEmpty { error "No reads matching: ${params.reads}" }
// .set { read_pairs_ch }
process align {
publishDir "${params.out}/aligned_reads", mode:'copy'
input:
set pair_id, file(reads) from read_pairs_ch
output:
set val(pair_id), file("${pair_id}_aligned_reads.sam") into aligned_reads_ch
script:
"""
module load bwa/intel/0.7.17
bwa mem \
$ref \
${reads[0]} \
${reads[1]} \
> ${pair_id}_aligned_reads.sam
"""
}
process sort_index_reads {
}
process aligned_read_stats {
}process {
executor = 'slurm'
cpus = 10
memory = '30 GB'
time = '30 min'
withName: align_reads { cpus = '20'}
}
Using sbatch to submit your workflow
#!/bin/bash #SBATCH –nodes=1 #SBATCH –ntasks-per-node=1 #SBATCH –cpus-per-task=2 #SBATCH –time=5:00:00 #SBATCH –mem=2GB #SBATCH –job-name=myJob #SBATCH –output=slurm_%j.out module purge module load jdk/1.8.0_111 module load nextflow/20.01.0 nextflow run main.nf -c your.config