Introduction
De novo (from new) genome assembly refers to the process of reconstructing an organism’s genome from smaller sequenced fragments. This is not a trivial task, and can involve multiple types of data and analysis methods/tools. A good analogy of this task is the example below Sequence Assembly Wiki.
The problem of sequence assembly can be compared to taking many copies of a book, passing each of them through a shredder with a different cutter, and piecing the text of the book back together just by looking at the shredded pieces. Besides the obvious difficulty of this task, there are some extra practical issues: the original may have many repeated paragraphs, and some shreds may be modified during shredding to have typos. Excerpts from another book may also be added in, and some shreds may be completely unrecognizable.
Typically, multiple paired end libraries are sequenced with short insert sizes (between 200bp-700bp) and these provide the bulk of sequencing coverage. Coverage can simply be computed by dividing the total number of sequenced bases by the “expected” size of the genome in question. This figure can vary according to the size and complexity of the organism, as well as the read length, but for smaller genomes (e.g. bacterial) 100x coverage is a good starting point. Long mate pair libraries are also commonly used in an effort to “bridge” assembled contigs/scaffolds and “stitch” the genome. These tend to have longer insert size (and opposite orientation to paired end libraries) ranging from 3kb-12kb. Coverage here is less of a concern since these are used mainly for scaffolding, so 20x coverage is usually a good recommendation. Finally, longer read length sequencing approaches (e.g. PacBio sequencing) can be used to fill in gaps in the assembled genome, as well as bridge contigs/scaffolds. Remember that while your mate pairs might help in stitching your genome, they will place gaps (stretches of N’s) between the bridged contigs/scaffolds. And whilst small and relatively “simple” genomes might assemble well using 1 paired end library, large, heterozygous, polyploid and repetitive genomes will require multiple rounds of sequencing using different libraries/technologies and potentially manual finishing of gaps.
In this tutorial, we will be assembling a bacterial genome that was sequenced using a standard paired end library approach. We will focus on assembling with 2 different tools, polishing these assemblies (gap closing), assessing them and selecting the best performing assembly, and finally annotating our genome and visualizing it.
There is no “best” approach, or a golden standard, when assembling a genome. Ideally, you want to use multiple tools/parameters and explore what best works in each individual case, and this can be a lengthy and sometimes complex analysis. This is why at NYUAD, we use dedicated analysis modules (in this case the gencore_de_novo_genome/1.0 module), to wrap multiple analysis tools in way that makes them easy to use, and ensures no compatibility issues between the different software (a problem that is very common in bioinformatics). We tend to couple our analysis with predefined and easily configurable workflows (yaml configuration files), which can execute the requested analysis and track parameters/inputs/outputs/dependencies from start to finish. Here, we will breakdown the individual commands and what some of the more commonly used arguments mean.
Overview
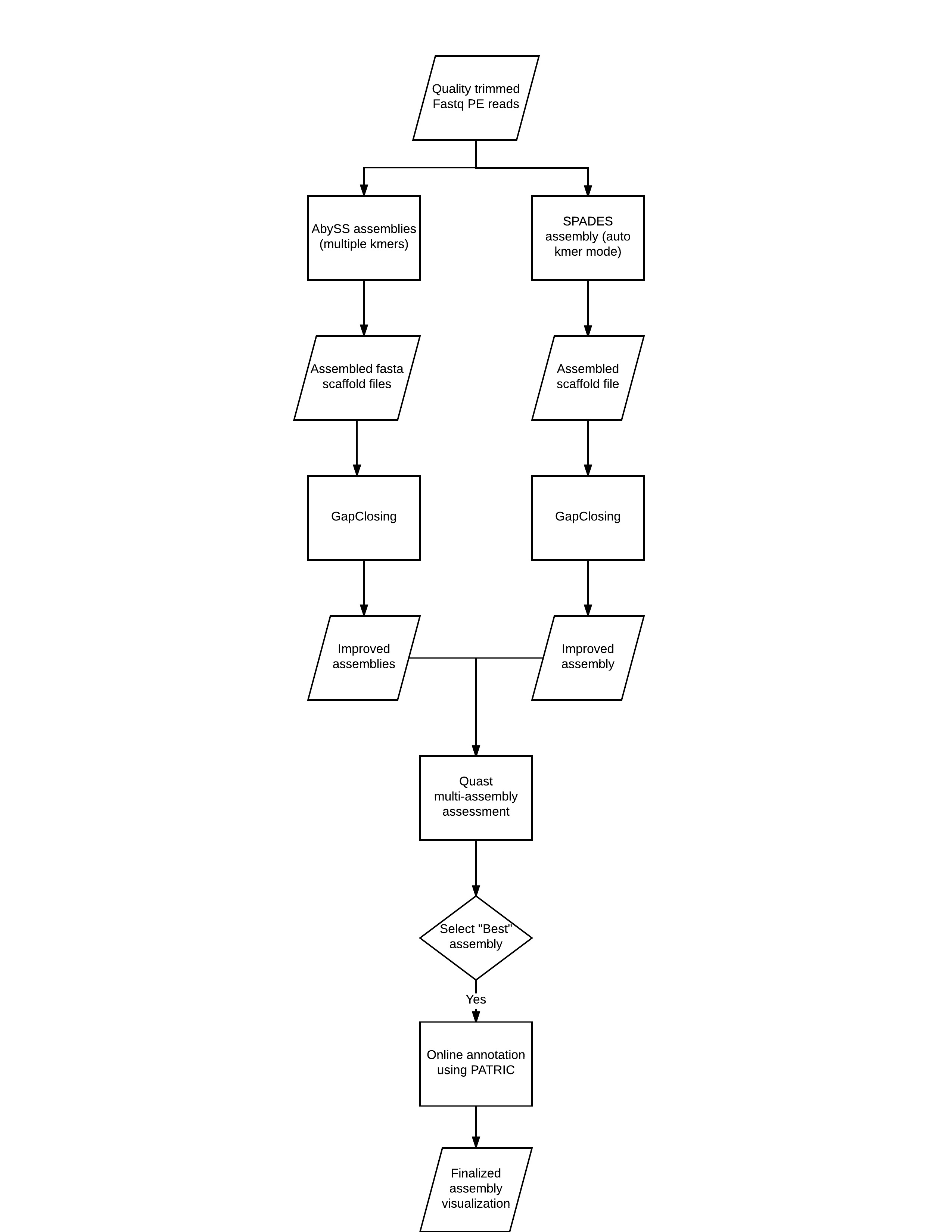
Of course, a complete de novo assembly will include multiple tools and parameters, including pre-assembly read error correction, kmer profiling, further scaffolding/gapclosing, among others. Below is a diagrammatic representation of a more comprehensive workflow.
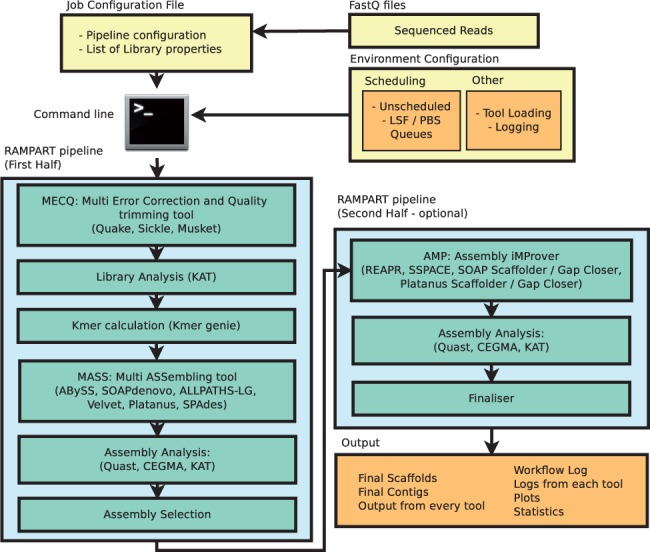
And remember that this is a short introduction to de novo genome assembly. Technologies and protocols, as well as analysis methods, are constantly evolving. So as always, do your research and stay up to date.
- RAMPART: a workflow management system for de novo genome assembly. Mapleson D, Drou N, Swarbreck D. 2015 Jun 1;31(11):1824-6. doi: 10.1093/bioinformatics/btv056. Epub 2015 Jan 30. ↩